南京大学学报(自然科学版) ›› 2022, Vol. 58 ›› Issue (1): 103–114.doi: 10.13232/j.cnki.jnju.2022.01.011
• • 上一篇
基于蛋白质序列的氨基酸字母表简化
张鑫鹏, 王骏( ), 王炜()
), 王炜()
- 南京大学物理学院,南京,210093
Simplification of amino acid alphabet based on protein sequences
Xinpeng Zhang, Jun Wang(), Wei Wang()
- School of Physics,Nanjing University,Nanjing,210093,China
摘要:
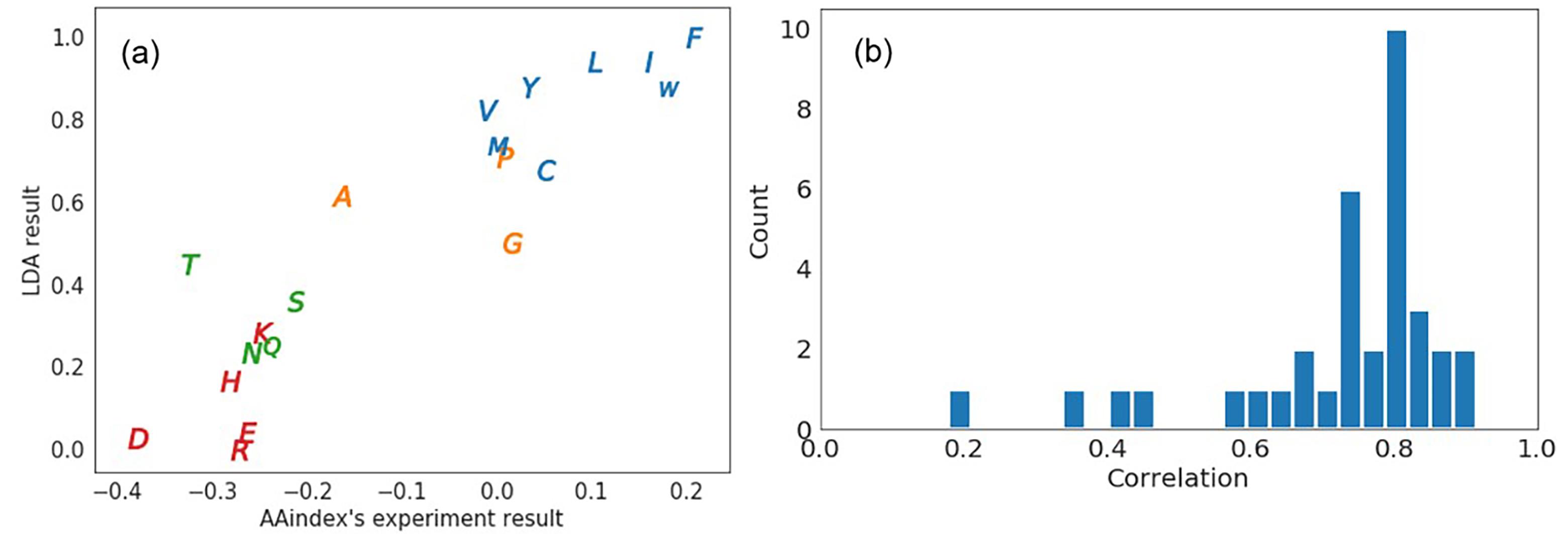
蛋白质的结构和功能特性由其氨基酸序列编码,控制序列结构映射的规则被认为是二级遗传密码,氨基酸字母表的简化可以减少蛋白质序列中的冗余,有助于揭示编码规则.基于氨基酸的单体特征、成对相互作用和相似性,可以简化氨基酸字母表.目前,仅基于蛋白质的序列信息,根据最近邻氨基酸的出现频率构建了一个氨基酸的嵌入表示.在此基础上,提出一种通过重构最近邻氨基酸的出现频率来压缩嵌入表示的模型,将此方法命名为AA2Vec.实验结果表明,与其他表示维相比,特定表示维(三维)具有显著的鲁棒性.提取的信息捕捉了氨基酸的物理化学和生化特性以及最近邻氨基酸之间的相互作用.值得注意的是,提出的方法对于具有不同序列标识的序列数据集(SCOPe)是稳定的.这种方法给出了氨基酸的最简表示,有助于生成蛋白质序列的简化表示和建立蛋白质的简化模型.
中图分类号:
- Q61















| 1 | Consortium UniProt. Uniprot:The universal protein knowledgebase in 2021. Nucleic Acids Research,2021,49(D1):D480-D489. |
| 2 | Senior A W,Evans R,Jumper J,et al. Improved protein structure prediction using potentials from deep learning. Nature,2020,577(7792):706-710. |
| 3 | Yang J Y,Anishchenko I,Park H,et al. Improved protein structure prediction using predicted interresidue orientations. Proceedings of the National Academy of Sciences of the United States of America,2020,117(3):1496-1503. |
| 4 | Xu J B,Mcpartlon M,Li J. Improved protein structure prediction by deep learning irrespective of co?evolution information. bioRxiv,2020,doi:10.1101/2020.10.12.336859. |
| 5 | Xu J B,Wang S. Analysis of distance?based protein structure prediction by deep learning in CASP13. Proteins:Structure,Function,and Bioinformatics,2019,87(12):1069-1081. |
| 6 | Hashemifar S,Neyshabur B,Khan A A,et al. Predicting protein?protein interactions through sequence?based deep learning. Bioinformatics,2018,34(17):i802–i810. |
| 7 | Wang S,Sun S Q,Li Z,et al. Accurate de novo prediction of protein contact map by ultra?deep learning model. PLoS Computational Biology,2017,13(1):e1005324. |
| 8 | Hanson J,Paliwal K K,Litfin T,et al. Getting to know your neighbor:Protein structure prediction comes of age with contextual machine learning. Journal of Computational Biology,2020,27(5):796-814. |
| 9 | Wang J,Wang W. Simplification of complexity in protein molecular systems by grouping amino acids:A view from physics. Advances in Physics:X,2016,1(3):444-466. |
| 10 | Cieplak M,Holter N,Maritan A,et al. Amino acid classes and the protein folding problem. The Journal of Chemical Physics,2001,114(3):1420-1423. |
| 11 | Pape S,Hoffgaard F,Hamacher K. Distance?dependent classification of amino acids by information theory. Proteins:Structure,Function,and Bioinformatics,2010,78(10):2322-2328. |
| 12 | Wang J,Wang W. A computational approach to simplifying the protein folding alphabet. Nature Structural Biology,1999,6(11):1033-1038. |
| 13 | Li T P,Fan K,Wang J,et al. Reduction of protein sequence complexity by residue grouping. Protein Engineering,2003,16(3):323-330. |
| 14 | Cannata N,Toppo S,Romualdi C,et al. Simplifying amino acid alphabets by means of a branch and bound algorithm and substitution matrices. Bioinformatics,2002,18(8):1102-1108. |
| 15 | Wrabl J Q,Grishin N V. Grouping of amino acid types and extraction of amino acid properties from multiple sequence alignments using variance maximization. Proteins:Structure,Function,and Bioinformatics,2005,61(3):523-534. |
| 16 | Asgari E,Mofrad M R K. Continuous distributed representation of biological sequences for deep proteomics and genomics. PLoS One,2015,10(11):e0141287. |
| 17 | Kimothi D,Soni A,Biyani P,et al. Distributed representations for biological sequence analysis. 2016,arXiv:. |
| 18 | Yang K K,Wu Z,Bedbrook C N,et al. Learned protein embeddings for machine learning. Bioinformatics,2018,34(15):2642-2648. |
| 19 | Hamid M N,Friedberg I. Identifying antimicrobial peptides using word embedding with deep recurrent neural networks. bioRxiv,2018,doi:10.1101/255505. |
| 20 | Rives A,Meier J,Sercu T,et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proceedings of the National Academy of Sciences of the United States of America,2021,118(15):e2016239118. |
| 21 | Murzin A G,Brenner S E,Hubbard T,et al. SCOP:A structural classification of proteins database for the investigation of sequences and structures. Journal of Molecular Biology,1995,247(4):536-540. |
| 22 | Sickmeier M,Hamilton J A,Legall T,et al. Disprot:The database of disordered proteins. Nucleic Acids Research,2007,35(S1):D786-D793. |
| 23 | Zhou Q,Tang P Z,Liu S X,et al. Learning atoms for materials discovery. Proceedings of the National Academy of Sciences of the United States of America,2018,115(28):E6411-E6417. |
| 24 | Hamilton W L,Leskovec J,Jurafsky D. Cultural shift or linguistic drift? Comparing two computational measures of semantic change∥Proceedings of 2016 Conference on Empirical Methods in Natural Language Processing. Austin,TX,USA:Association for Computational Linguistics,2016:2116-2121. |
| 25 | Artetxe M,Labaka G,Agirre E. Learning principled bilingual mappings of word embeddings while preserving monolingual invariance∥Proceedings of 2016 Conference on Empirical Methods in Natural Language Processing. Austin,TX,USA:Association for Computational Linguistics,2016:2289-2294. |
| 26 | Smith S L,Turban D H P,Hamblin S,et al. Offline bilingual word vectors,orthogonal transformations and the inverted softmax. 2017,arXiv:. |
| 27 | Henikoff S,Henikoff J G. Amino acid substitution matrices from protein blocks. Proceedings of the National Academy of Sciences of the United States of America,1992,89(22):10915-10919. |
| 28 | Kawashima S,Pokarowski P,Pokarowska M,et al. AAindex:Amino acid index database,progress report 2008. Nucleic Acids Research,2008,36(S1):D202-D205. |
| 29 | Black S D,Mould D R. Development of hydrophobicity parameters to analyze proteins which bear post?or cotranslational modifications. Analytical Biochemistry,1991,193(1):72-82. |
| [1] | 潘越,王骏,李文飞,张建,王炜. 基于卷积神经网络的蛋白质折叠类型最小特征提取[J]. 南京大学学报(自然科学版), 2020, 56(5): 744-753. |
| [2] | 李佳云,吴人杰. 基因转录爆发的建模研究[J]. 南京大学学报(自然科学版), 2020, 56(3): 418-429. |
|
||